
- UPDATES
-

-

-






Thinking, Sensing & Behaving



Diseases & Disorders

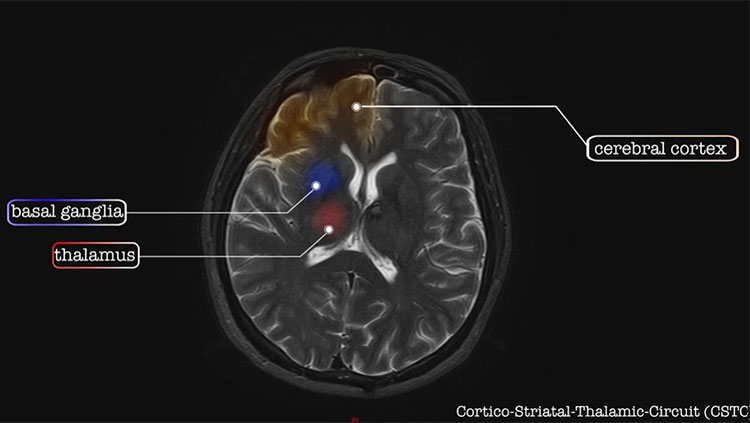
Brain Anatomy & Function
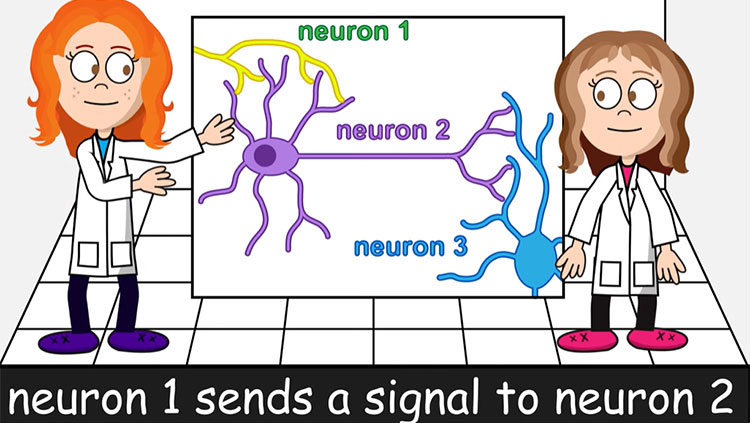
3D Brain
An interactive brain map that you can rotate in a three-dimensional space.
Neuroscience in Society


In the Lab


