Physiology of Liquefactive Necrosis Suggests Treatments for Atherosclerosis Could Potentially Be Applied to Stroke
This article was co authored by Matthew Davidson, PhD and eNeuro Features Editor Rosalind S.E. Carney, DPhil. Dr. Davidson is currently the Science Policy Fellow at Research!America. Follow them on Twitter at @MLDSciWizard
In their eNeuro publication, Chung, Frye, and colleagues examined why the heart and brain exhibit different responses to ischemia. Following ischemia induced by myocardial infarction, cardiac tissue undergoes coagulative necrosis, a chronic inflammatory response that persists for approximately four weeks. In contrast, stroke-induced ischemia causes an inflammatory response lasting several months in which brain tissue progressively degenerates into a liquid mass in a process called liquefactive necrosis. It was not known why ischemia results in differences in inflammatory response duration or mechanisms of tissue degeneration in the heart and brain. The authors found that stroke-induced ischemia shares some physiological characteristics with atherosclerosis, including the formation of cholesterol crystals. These results suggest that treatments for atherosclerosis could potentially be applied to stroke.
The high cholesterol levels associated with atherosclerosis overwhelm the phagocytic capability of macrophages, leading to cholesterol deposition in blood vessels and the restriction of blood flow. Of all the human organs, the brain has the highest cholesterol content, predominantly as a component of myelin. A prior study by others showed that myelin debris can lead to chronic inflammation and the formation of cholesterol crystals in the brains of aged mice. Therefore, Chung, Frye, and colleagues hypothesized that following stroke, the high quantity of damaged myelin sheaths could result in excessive cholesterol levels that cause the prolonged inflammatory response observed in liquefactive necrosis.
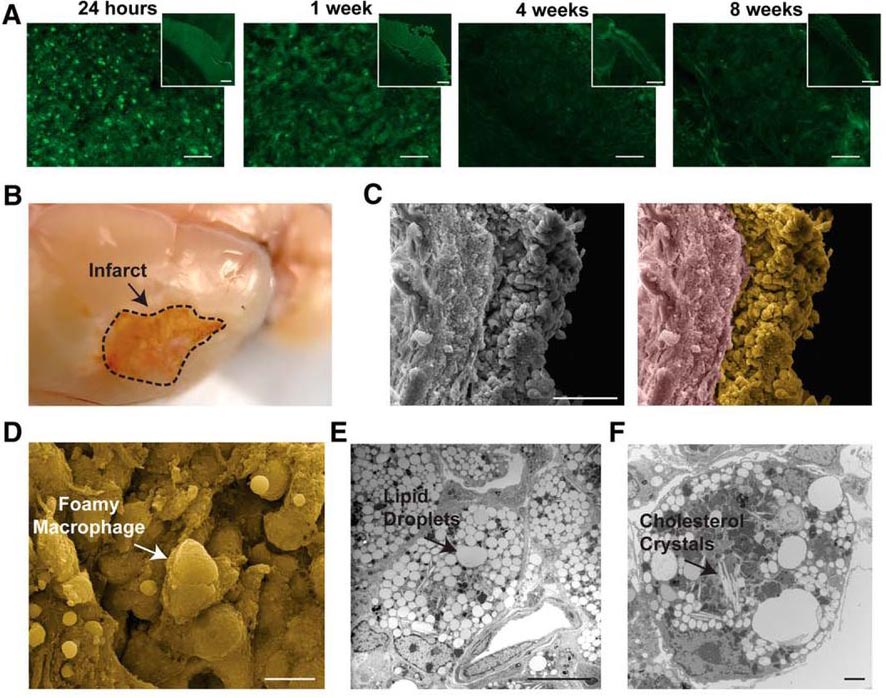
The authors examined the changes in gross morphology that occur in a mouse model of experimentally induced stroke. Use of a fluorochrome that visualizes degenerating neurons revealed that neuronal degeneration was visible within 24 hours post-ischemia and continued until the final time point examined, at eight weeks post-ischemia (Figure 1). The infarct site had transformed into liquefied tissue that contained foamy macrophages, lipid droplets, and cholesterol crystals (Figure 1; shown at seven weeks post-ischemia). As these observations were consistent with the pathology of atherosclerotic plaques, these findings lent credence to the hypothesis of a link between liquefactive necrosis and atherosclerosis.
Figure 1. Morphological pathology of liquefactive necrosis up to eight weeks post-ischemia in a mouse model of stroke. (A) Degenerating neurons are observed at the infarct site 24 h, 1 week, 4 weeks, and 8 weeks post-ischemia. (B) At 7 weeks post-ischemia, the infarct site exhibits liquified brain tissue. (C) The area of liquefaction contains a dense population of cells adjacent to the glial scar as imaged by scanning electron microscopy. In the right image, the area of liquefaction is pseudocolored yellow and the glial scar is pseudocolored purple. (D-E) The infarct site contains foamy macrophages (D), lipid droplets (E), and cholesterol crystals (F). (Adapted from Figure 3 in Chung et al. 2018).
Cholesterol crystals were not observed at any time-point post-ischemia in the heart in a mouse model of myocardial infarction. The appearance of cholesterol crystals in the brain coincided with a secondary wave of inflammation four to eight weeks post-ischemia, characterized by increased expression levels of several pro-inflammatory cytokines, including MCP-1, IP-10, and RANTES. These results indicated that brain liquefaction includes a biphasic cytokine response. A prior study by others showed that regression of atherosclerosis was achieved by inhibiting pro-inflammatory cytokines, such as MCP-1, IP-10, or RANTES, again providing further support for the potential use of treatments for atherosclerosis to be applied to stroke.
The secondary wave of inflammation in the brain also correlated with increased expression levels of the secreted protein osteopontin (OPN); upregulation of OPN is also observed in atherosclerosis. OPN modulation is another potential target for treatment following stroke because the authors also show that OPN knockout (OPN–/– ) mice had lower levels of proinflammatory cytokines and matrix metalloproteinases, less neurodegeneration, and faster recovery of motor function. However, OPN–/– mice also had higher levels of cholesterol crystals in the stroke-induced infarct. Chung, Frye, and colleagues hypothesize that OPN deficiency reduces the clearance rate of myelin debris, therefore enabling the formation of cholesterol crystals. Therefore, further research is needed to determine whether the observed benefits of OPN deficiency are negated by the additional cholesterol crystal formation.
Until this eNeuro publication, it was not clear why brain tissue undergoes liquefaction or why inflammation lasts so long compared to cardiac tissue. The results of this study indicate that the high cholesterol content of the brain underlies its prolonged and severe degeneration.
Read the full article:
Liquefaction of the Brain following Stroke Shares a Similar Molecular and Morphological Profile with Atherosclerosis and Mediates Secondary Neurodegeneration in an Osteopontin-Dependent Mechanism.
Amanda G. Chung*, Jennifer B. Frye*, Jacob C. Zbesko, Eleni Constantopoulos, Megan Hayes, Anna G. Figueroa, Danielle A. Becktel, W. Antony Day, John P. Konhilas, Brian S. McKay, Thuy-Vi V. Nguyen and Kristian P. Doyle *indicates co-authors
FOLLOW US
 RSS Feed
RSS FeedPOPULAR POSTS
TAGS
CATEGORIES




