Chaperoning the Rescue of Synaptic Vesicle Recycling Defects Caused by Excess Alpha-Synuclein
Synucleinopathies, such as Parkinson’s disease, are a type of neurogenerative disorder associated with excessive accumulation of alpha-synuclein (α-synuclein) in insoluble cytoplasmic aggregates called Lewy Bodies. Within a healthy neuron, normal levels of α-synuclein expression are predominantly observed in the presynapse. The presynapse contains synaptic vesicles which store neurotransmitters until an action potential results in neurotransmitter release into the synaptic cleft. This neurotransmitter release event activates receptors on the postsynaptic neurons allowing neurons to communicate with each other. To ensure that subsequent neurotransmitter release can occur rapidly, components of synaptic vesicles and neurotransmitters are taken back into the presynapse for recycling. Several forms of endocytosis can facilitate the recycling of vesicle components from the plasma membrane into the presynapse to enable the formation of new synaptic vesicles. Clathrin-mediated endocytosis (CME) is thought to be the predominant mechanism relevant to the model organism and synapse investigated by Banks, Medeiros, and colleagues. Previous research from Associate Scientist Jennifer Morgan’s laboratory (Marine Biological Laboratory, Woods Hole, MA) showed that when excess α-synuclein is present at the presynapse, synaptic vesicle recycling is impaired. The observations were consistent with a disruption in CME as demonstrated by a reduced number of internalized synaptic vesicles, an expansion of the plasma membrane, as well as increased numbers of clathrin-coated intermediates that will be described below (Busch et al., 2014). Given that several inherited forms of Parkinson’s Disease involve multiplication of the gene that encodes α-synuclein, resulting in increased levels of α-synuclein protein, it is crucial to understand the molecular mechanism by which excess α-synuclein impairs the synaptic vesicle recycling process mediated by CME.
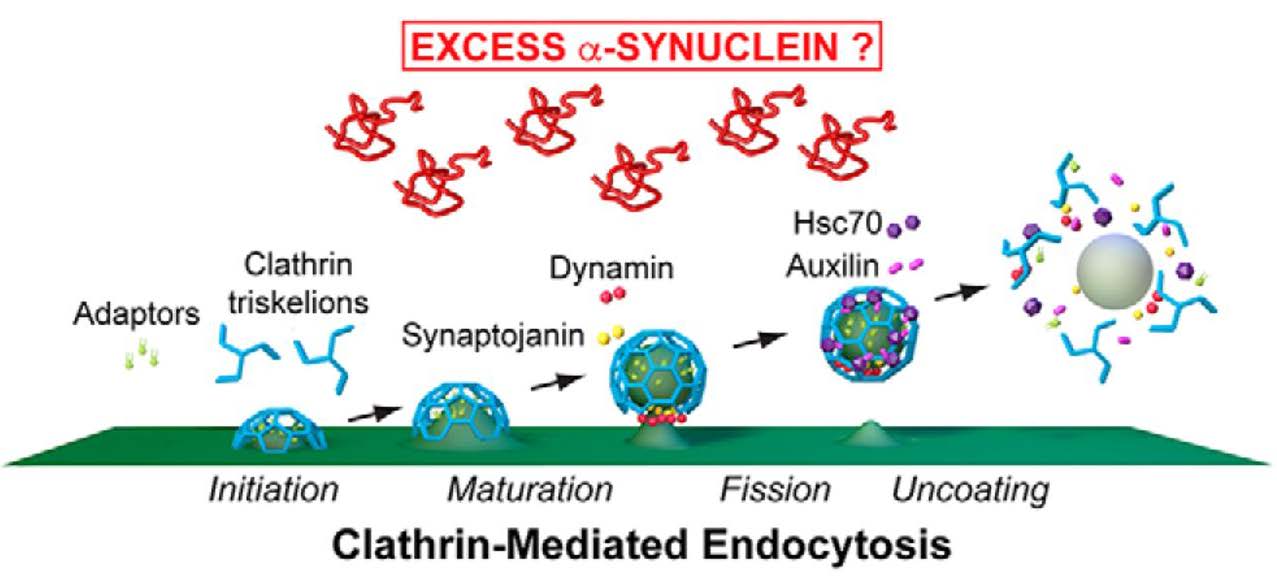
Figure 1 shows a schematic of the steps of CME during which excess α-synuclein could cause the synaptic vesicle recycling process to become defective. CME includes initiation, maturation, fission, and uncoating stages of the synaptic vesicle recycling process. CME commences when clathrin adaptor proteins target 3-legged clathrin units, called triskelia, to the plasma membrane. The triskelia self-assemble into a clathrin coat, which forms a cage-like invagination of the plasma membrane called a clathrin-coated pit (CCP). The CCP develops into a clathrin-coated vesicle (CCV), which is separated from the plasma membrane via the GTPase activity of dynamin. The CCV, now internalized within the presynapse, undergoes a clathrin-uncoating process mediated by the chaperone protein Hsc70 and its co-chaperone auxilin. Uncoated vesicles can then incorporate neurotransmitters to become primed again for neurotransmitter release. The electron-dense coat of CCPs and CCVs facilitates visualization of these intermediate synaptic recycling structures in electron microscopy (Morgan et al., 2013). Banks, Medeiros, and colleagues used ultrastructural analyses to identify the molecular target(s) of the CME process impaired by excess α-synuclein.
The sea lamprey (Petromyzon marinus)is a jawless fish that, although primitive, is a good vertebrate model organism in which to study neurotransmission. Neurons of the lamprey spinal cord are individually distinct, neuroanatomically defined, easily accessible for manipulation, and large enough to permit ultrastructural analyses. The authors focused their research on the giant reticulospinal (RS) synapse, a classical glutamatergic vertebrate synapse, that signals via the same molecules as mammalian neurotransmission. The large size of RS axons enables proteins to be microinjected locally at the presynapse, resulting in an acute perturbation independent of the compensatory effects that can follow experimental manipulations such as chronic protein overexpression. In addition, because they have very large synaptic vesicle clusters (1000–2000 vesicles), RS synapses provide greater ultrastructural detail than is possible with most rodent models. The authors electrically stimulated each lamprey axon (5 minutes at 20 Hz) immediately prior to fixation and processing for electron microscopy. Synapses within the same axon served as internal controls—these axons were electrically stimulated but not microinjected. The ability to use internal controls was important as synaptic vesicles exhibit not only inter-animal variability but also intra-animal variability, depending on the axon.
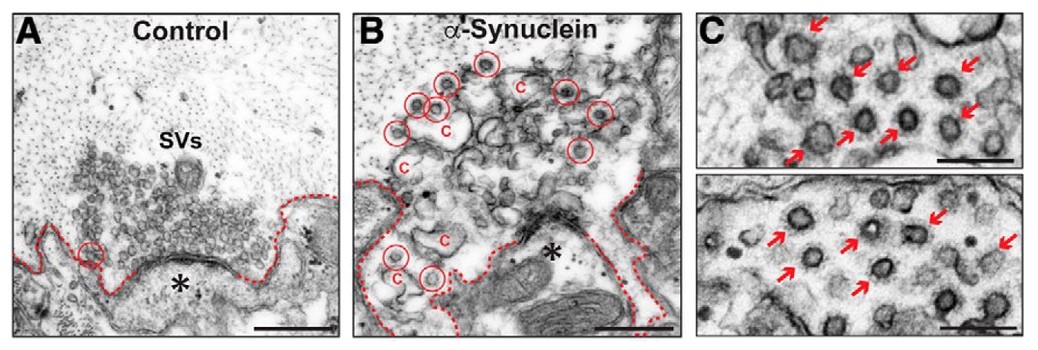
Control synapses exhibited a large number of synaptic vesicles and few CCPs/CCVs (synaptic recycling intermediates), indicative of efficacious CME (Figure 2A). The authors microinjected human α-synuclein to a final axonal concentration 2–4 times greater than endogenous levels to mimic overexpression levels observed in individuals with Parkinson’s Disease. As expected, excess human α-synuclein at the presynapse resulted in impaired CME evidenced by fewer synaptic vesicles and greater numbers of free CCVs (Figure 2B,C). These results suggest that the presence of excess human α-synuclein at the presynapse results in a specific defect in CCV uncoating during CME-mediated synaptic vesicle recycling.
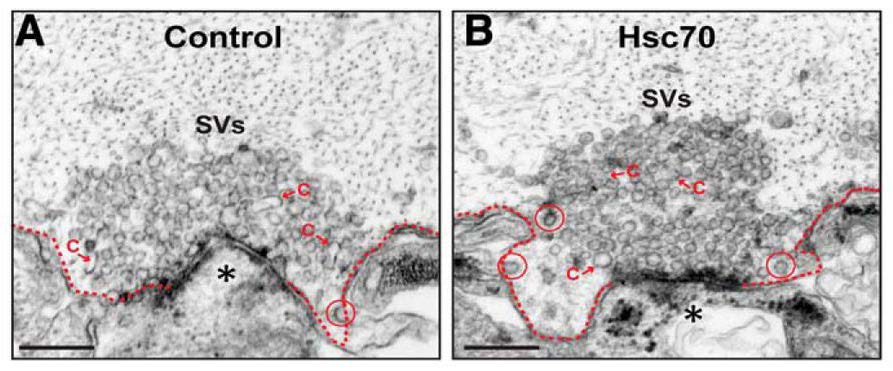
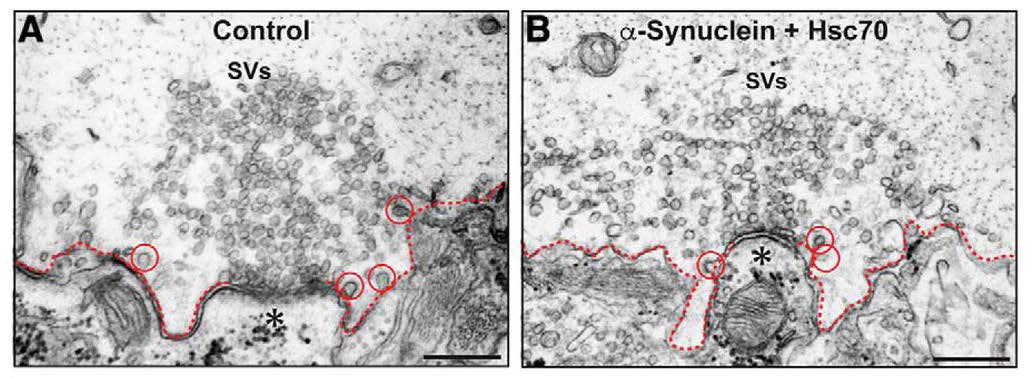
To identify which molecular component of the CME process is affected by excess human α-synuclein, the authors performed biochemical pull-down assays. Binding of human α-synuclein or lamprey synuclein to Hsc70 was observed. This observation, in addition to the ultrastructural analyses, implicated the CCV uncoating step as the timepoint of CME that is most likely disrupted by excess α-synuclein. Immunofluorescence analysis showed that the normal increase in Hsc70 availability at synapses did not occur in electrically stimulated synapses that had excess human α-synuclein. This observation suggested that Hsc70 availability at the synapse may underlie the defects in CME caused by excess human α-synuclein. Banks, Medeiros, and colleagues found that introduction of excess Hsc70 alone did not induce a CME phenotype compared to untreated controls (Figure 3). However, exogenous Hsc70 administered in combination with excess human α-synuclein rescued the prior CME phenotype found with the introduction of only excess human α-synuclein at the presynapse (Figure 4). Specifically, the human α-synuclein + Hsc70 condition resulted in large synaptic vesicle clusters and normal numbers of CCPs/CCVs. These results identified Hsc70 as the molecular target of excess human α-synuclein-associated defects in CME. Importantly, the authors show that the aberrant CME phenotype can be rescued in vivo by microinjection of the chaperone protein that promotes CCV uncoating.
CME is also the process by which transferrin, low-density lipoprotein, and peptide hormones are endocytosed into cells. In the neuron, AMPA receptors are internalized into dendrites (receptor-mediated endocytosis), which facilitates synaptic plasticity. Therefore, it will be beneficial to broaden research into how excess α-synuclein affects the uptake of molecules at other locations within neurons and in other cells. Interestingly, Hsc70 is also a major component of Lewy bodies, suggesting altered chaperone function in other neuronal compartments. Banks, Medeiros, and colleagues’publication is an advance in the field because they have identified molecular components of CME that are affected by the presence of excess human α-synuclein at a glutamatergic synapse. The results are particularly compelling in that the authors introduced an acute perturbation into a normally functioning synapse in vivo and identified a molecular target that causes an aberrant phenotype. More defined relevance for Parkinson’s Disease would involve applying these findings to overexpression studies in other model organisms in and further investigating defective synaptic neurotransmission in Parkinson’s Disease and other synucleinopathies.
References:
- Busch DJ, Oliphint PA, Walsh RB, Banks SML, Woods WS, George JM, et al. Acute increase of α-synuclein inhibits synaptic vesicle recycling evoked during intense stimulation.Mol Biol Cell. 2014 Dec 1;25(24):3926–41.
- Morgan JR, Jiang J, Oliphint PA, Jin S, Gimenez LE, Busch DJ, et al. A Role for an Hsp70 Nucleotide Exchange Factor in the Regulation of Synaptic Vesicle Endocytosis.J Neurosci.2013 May 1;33(18):8009–21.
Read the full article:
Hsc70 Ameliorates the Vesicle Recycling Defects Caused by Excess α-Synuclein at Synapses
Susan M. L. Banks, Audrey T. Medeiros, Molly McQuillan, David J. Busch, Ana Sofia Ibarraran-Viniegra, Rui Sousa, Eileen M. Lafer, and Jennifer R. Morgan
FOLLOW US
 RSS Feed
RSS FeedPOPULAR POSTS
TAGS
CATEGORIES




